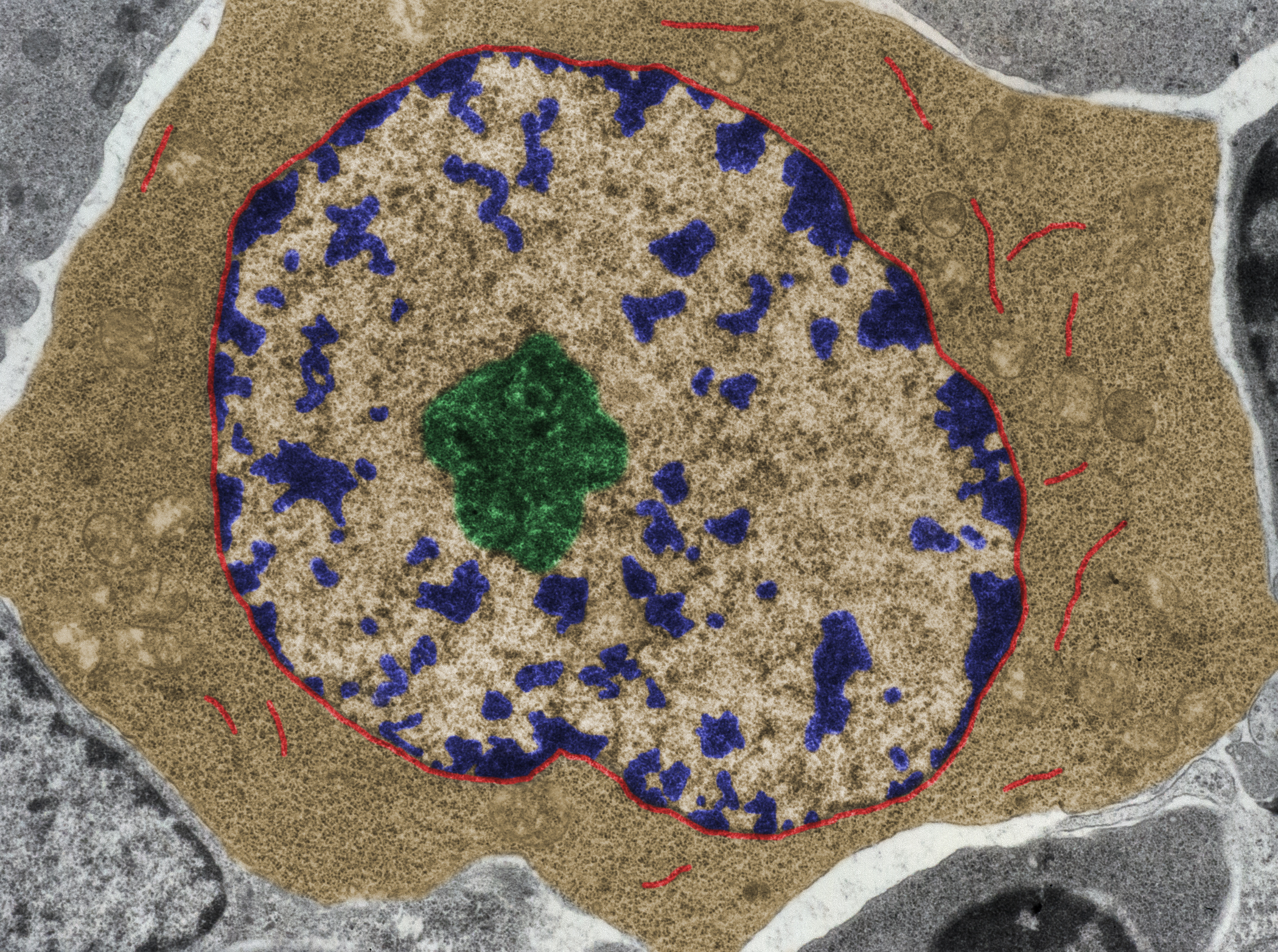
For instance, the chromatin, which is the large molecule that forms a chromosome, can be tightly wrapped up in certain areas, but it can be open in other areas. And guess where the genes are being expressed? In the areas that are open. The genes that are in the areas that are closed are not able to be expressed, and that’s how cells differentiate. In tumours, you kind of recapitulate the entire human development. And so just understanding how the epigenetics of the cells were complemented by gene expression and mutation, etc. gave us a set of data that fostered the development of the field.
Developmental biologists are now starting to generate the same type of data from all sorts of different layers of lineage development in the human body or in the body of model animals. Many disciplines to do with neurobehavioural or neurodegenerative diseases are starting to generate the same wealth of information. But cancer was the first one. And thanks to those efforts, we were able to start developing the very first framework for the computational analysis of biological data.